

À época eu trabalhava com reações de oxiarilação catalizada por paládio e então escolhi um tema que me agradava (e ainda agrada) e que fazia parte do meu trabalho, a reação de Heck. O foco era nos aspectos regioquímicos dessa reação sensacional! A seguir vocês encontrarão o que consistia o manuscrito, que na verdade é uma revisão da literatura com uma abordagem didática.
I- A Reação de Heck:
A formação de novas ligações carbono-carbono (C-C) é o maior desafio em qualquer planejamento sintético,[1] é etapa chave em várias sínteses de substâncias orgânicas e de produto naturais, possuindo varias aplicações industriais e tecnológicas.[2]
Das ferramentas sintéticas disponíveispara formação de novas ligações C-C podemos destacar o importante papel da catálise organometálica, [3] em especial as
reações mediadas por complexos organometálicos de paládio, [4] destes processos a reação de Heck possui papel de destaque. [4-8]
A reação de Heck consiste na arilação ou vinilação de olefinas, na presença de complexos de paládio e uma base. [4, 5, 9] (Esquema 2) Foi primeiramente descrita independentemente por Mizoroki [10] e por Heck [11-13] na década de 60, onde os primeiros experimentos eram realizados na presença de organomercuriais e quantidades estequiométricas de paládio. (Esquema 1) Posteriormente foram desenvolvidas metodologias utilizando paládio em quantidades catalíticas e substituindo-se os organomercuriais por haletos de arila ou vinila.[9, 14, 15]
Outros eletrófilos além dos haletos também podem ser empregados, como triflatos, sais de diazônio, cloretos de ácido, anidridos, etc.[4] Na versão catalítica, é muito comum o emprego de ligantes a fim de estabilizar os complexos de paládio formados, dentre esses ligantes as fosfinas são as mais amplamente empregadas.[6, 7, 9, 16,17]


Grande variedade de grupos funcionais é tolerado na reação de Heck, o que a torna extremamente versátil.[4, 6, 9]
II- Mecanismo da reação de Heck:
O ciclo catalítico envolve 4 reações definidas e diversos intermediários (Figura 1):
Primeiro, após a geração da espécie ativa do catalisador de paládio (Pd(0)) ocorre a adição oxidativa do substrato eletrofílico RX na espécie ativa de Pd(0), gerando o aduto RPdL2X.[6, 7, 9, 17, 18]
Segundo, liberação de um sítio de ligação (L ou X), e subseqüente coordenação da olefina resultando num complexo-π que pode ser neutro ou positivamente carregado, dependendo de qual tipo de ligante foi liberado previamente para coordenação da olefina.
Terceiro, ocorre uma inserção syn (carbopaladação), provavelmente via um mecanismo consertado e estado de transição de quatro centros, resultando na geração de um complexo instável σ−organopaládio, é nesta etapa que é determinada a regioquímica do produto da reação. [6, 7, 9, 17]
Quarto, um hidrogênio β torna-se acessível através de uma rotação interna, colocando-se numa posição syn ao metal, este perde um ligante gerando agora um sítio de coordenação para que ocorra a eliminação β de hidreto seguida da liberação do produto de acoplamento de Heck e hidreto de paládio.[4, 6, 7, 9, 17, 19]
Por último, é necessário uma base para a reciclagem do hidreto de paládio (Pd(II)) a espécie ativa (Pd(0)) para reinício do ciclo catalítico. (Figura 1)[6, 7, 9, 17]

III- Mecanismo neutro X mecanismo catiônico (polar)
Na etapa de complexação/inserção da olefina, é necessário o prévio desligamento de um ligante da esfera de coordenação do paládio para que ocorra a interação com a olefina.
Se o ligante a se desligar for um ligante neutro (ligantes de fosfina, por exemplo) o complexo formado será neutro (o haleto permanece ligado ao metal). Caso o ligante a ser liberado seja um ligante do tipo aniônico (como os haletos, por exemplo) o complexo formado será do tipo catiônico (ou polar), esse tipo de intermediário ocorre quando no meio reacional existe um sequestrador de haletos, como sais de prata, tálio
ou titânio;[6, 20, 21] ou ainda quando triflatos ou sais de diazônio são usados como eletrófilos.[17, 22]
(Esquema 3)
O mecanismo catiônico também pode ser favorecido por ligantes bidentados, que são amplamente utilizados na versão assimétrica da reação de Heck.[9]

IV- Regioquímica da etapa de inserção:
De maneira geral a reação se processa facilmente com olefinas terminais que possuam grupos puxadores de elétrons. No entanto, olefinas eletronicamente neutras e com grupos doadores de elétrons são substratos menos adequados.[5, 6, 22] Com olefinas ricas em elétrons, nas condições usuais de Heck, são obtidos misturas de produtos de mono e di-arilação, e misturas de regioisômeros.[17, 22, 23] (Esquema 4)

Contudo, métodos eficientes foram desenvolvidos, tornando possível a funcionalização de olefinas ricas em elétrons de maneira regiocontrolada, bem como também o controle da migração da dupla ligação em sistemas cíclicos e acíclicos.[6, 22-27]
As diferentes condições reacionais resultam em mecanismos neutro ou catiônico (polar), e a seletividade da reação de Heck dependerá tanto do tipo de grupo abandonador, como também da densidade eletrônica do sistema vinílico, como pode ser observado nos estudos competitivos realizados por Cabri e colaboradores.[26] Complexos neutros reagem mais rápido com olefinas pobres em elétrons enquanto complexos catiônicos reagem mais rápido com olefinas ricas em elétrons.[22] (Esquema 5)
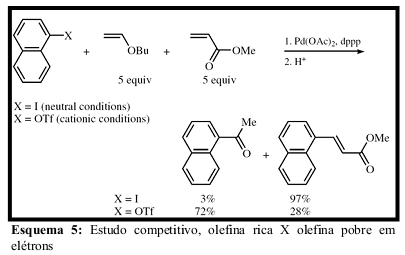
Cabri e colaboradores utilizando ligantes bidentadose condições que levam à reação por mecanismo neutro ou catiônico, demonstraram que olefinas com demandas eletrônicas diferentes podem apresentar regioquímica de arilação diferente dependendo das condições reacionais empregadas.[22] (Figura 2)

Podemos observar que olefinas deficientes em elétrons possuem o mesmo comportamento em ambos os mecanismos, o produto obtido é o produto linear (arilação na posição β), neste caso fatores estéricos controlam a regioquímica da etapa de inserção sendo preferencial a arilação no carbono menos estericamente impedido.[22]
Nestas condições olefinas ricas em elétrons apresentam comportamento diferente. Sob mecanismo neutro, ocorre mistura de produto linear e ramificado (arilação βe α) enquanto sob condições de mecanismo catiônico ocorre formação preferencial do produto ramificado (arilação α) devido a maior polarização da olefina no complexo πcarregado, neste caso o balanço de efeitos estéricos e eletrônicos
controlam a etapa de inserção sendo preferencial a arilação no carbono com menor
densidade eletrônica.[22]
O modelo de Dewar-Chatt-Duncanson de interação olefina-metal pode explicar a maior polarização de olefinas ricas em elétrons em complexos catiônicos. Olefinas ricas em elétrons são fracas receptoras πe boas doadoras σ, enquanto olefinas pobres são boas receptoras π e fracas doadoras σ.[28−31] (Figura 3)

Cenário semelhante é descrito por Anderson e Hallberg para explicar a seletividade da arilação na posição αem enol éteres,[32] onde a interação do HOMO da olefina com o orbital antiligante (σ*) da ligação aril-paládio(II) define a regioquímica do produto (Figura 4).

A regioseletividade da reação de Heck é determinada pela diferença de energia relativa entre os dois caminhos possíveis de inserção (ΔΔE=ΔE*(α)-ΔE*(β)),[33] fatores entrópicos que favoreçam um ou outro estado de transição influenciarão na regioquímica do produto.[34]
Podemos destacar o estireno, que apresenta seletividade para arilação na posição αem ambos caminhos reacionais (neutro ou catiônico). Ludwig e colaboradores,[35] demonstraram através de P31 RMN e por cálculos de modelagem molecular que essa seletividade deve-se a estabilização do tipo η3 π-benzil do intermediário de inserção que possui o átomo de paládio na posição benzílica (Figura 5)

Efeitos de quelação também podem influenciar a regioquímica da etapa de inserção, como pode se observado no Esquema 6.

No entanto, na presença da fosfina bidentada o cenário muda, sendo o sitio de coordenação liberado não acessível ao grupo amino, sendo neste caso o produto ramificado formado (arilação β).[6]
A síntese do antagonista do receptor de dopamina, Preclamol [(-)-3-PPP] (Figura 6), utiliza abordagem semelhante para arilação seletiva na posição 2 do anel l-propyl-1,2,3,6-tetrahydropiridina (Esquema 7).[36]


Quando Ω é negativo o carbono βé atacado, levando ao produto linear, enquanto quando Ω é positivo o carbono αé atacado, formando o produto ramificado.[34]
Quando Ω é próximo de zero, mistura de produtos é esperada, e comumente observado (Tabela 1).[34]
Os valores de Ωsão aditivos podendo prever a regioquímica da adição em olefinas disubstituidas (Figura 7).[34]


V- Regioseletividade na etapa eliminação β e migração da dupla ligação.
O desenvolvimento da reação de Heck intermolecular em sistemas cíclicos começou a ser desenvolvida na década de 1970. Imediatamente o problema da reversibilidade da eliminação βde hidreto tornou-se evidente, complicando o uso em olefinas endocíclicas e em cadeias alifáticas longas, nestes casos misturas de isômeros eram obtidos.[6, 7, 9] (Esquema 8)


Outro exemplo é a arilação de álcoois homoalílicos, onde a presença ou não de sais de prata, determina o produto formado (Esquema 10).[38]

O efeito mediado pela prata deve-se a formação do complexo hidreto de paládio catiônico que é menos acessível para realizar a reinserção no produto de arilação.[6]
A reação de Heck em sistemas cíclicos é uma poderosa ferramenta na formação de centros estereogênicos. Como exemplo, podemos citar a síntese de um promissor inibidor do fator de agregação plaquetária (PAF), onde duas arilações sucessivas no sistema diidrofurano, utilizando condições neutras seguido de catiônicas geram o produto de esterioquímica relativa trans, que posteriormente é hidrogenado gerando o produto final (Esquema 11).[39]


VI- Conclusão:
Dados teóricos e experimentais tornam possível delinear algumas previsões quanto à regioquímica da reação de Heck, no entanto não podem ser tomados como absolutos, uma vez que esses estudos são limitados a sistemas relativamente simples.
Não é incomum em sistemas mais complexos essas previsões falharem, contudo a escolha de condições experimentais adequadas podem selecionar o produto desejado.
A reação de Heck indiscutivelmente é uma das mais poderosas ferramentas sintéticas da síntese orgânica moderna.
VII- Referências
2. Herrmann, W.A., V.P.W. Bohm, and C.P. Reisinger,Journal of Chemical Education, 2000. 77(1): p. 92
3. Schwartz, J. and J.A. Labinger,Journal of Chemical Education, 1980. 57(3): p. 170-175.
4. Beletskaya, I.P. and A.V. Cheprakov,Chemical Reviews, 2000. 100(8): p. 3009-3066.
5. Demeijere, A. and F.E. Meyer,Angewandte Chemie-International Edition, 1995. 33(23-24): p. 2379
6. E.Negishi, Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1&2. 2002: John Wiley & Sons, Inc. 3313.
7. F. Diederich and P.J.Stang, Metal-Catalyzed Cross-Coupling Reaction. 1 ed. 1998, Ney York: Wiley-VCH.
8. Negishi, E.,Journal of Organometallic Chemistry, 1999. 576(1-2): p. XV-XVI.
9. Tsuji, J., Palladium Reagents and Catalysts Innovations in Organic Synthesis 1995: JOHN WILEY & SONS
10. Mizoroki, T., K. Mori, and A. Ozaki,Bulletin of the Chemical Society of Japan, 1971. 44(2): p. 581.
11. Heck, R.F.,Journal of the American Chemical Society, 1968(90): p. 5518.
12. Heck, R.F.,Journal of the American Chemical Society, 1968(90): p. 5526.
13. Heck, R.F.,Journal of the American Chemical Society, 1968(90): p. 5531.
14. Heck, R.F. and J.P. Nolley,Journal of Organic Chemistry, 1972(32): p. 230.
15. Heck, R.F. and H.A. Dieck,Journal of the American Chemical Society, 1974(96): p. 1136.
16. Amatore, C., A. Jutand, and M.A. Mbarki,Organometallics, 1992. 11(9): p. 3009-3013.
17. Knowles, J.P. and A. Whiting,Organic & Biomolecular Chemistry, 2007. 5(1): p. 31-44.
18. Amatore, C. and A. Jutand,Journal of Organometallic Chemistry, 1999. 576(1-2): p. 254-278.
19. Schmidt, A.F. and V.V. Smirnov,Kinetics and Catalysis, 2003. 44(4): p. 518-523.
20. Sato, Y., M. Sodeoka, and M. Shibasaki,Chemistry Letters, 1990(10): p. 1953-1954.
21. Karabelas, K., C. Westerlund, and A. Hallberg,Journal of Organic Chemistry,
1985. 50(20): p. 3896-3900.
22. Cabri, W. and I. Candiani,Accounts of Chemical Research, 1995. 28(1): p. 2-7.
23. Crisp, G.T.,Chemical Society Reviews, 1998. 27(6): p. 427-436.
24. Cabri, W., et al.,Journal of Organic Chemistry, 1992. 57(5): p. 1481-1486.
25. Cabri, W., et al.,Journal of Organic Chemistry, 1990. 55(11): p. 3654-3655.
26. Cabri, W., et al.,Journal of Organic Chemistry, 1992. 57(13): p. 3558-3563.
27. Zhang, Z.H., et al.,Journal of Organic Chemistry, 2006. 71(11): p. 4339-4342.
28. Chatt, J. and L.A. Duncanson,Journal of the Chemical Society, 1953(OCT): p. 2939-2947.
29. Frenking, G. and N. Frohlich,Chemical Reviews, 2000. 100(2): p. 717-774.
30. Crabtree, R.H., The Organometallic Chemistry Of The Transition Metals. Fourth Edition ed. 2005: John Wiley & Sons.
31. McGuinness, D.S., et al.,Organometallics, 1999. 18(9): p. 1596-1605.
32. Andersson, C.M., A. Hallberg, and G.D. Daves,Journal of Organic Chemistry, 1987. 52(16): p. 3529
33. von Schenck, H., B. Akermark, and M. Svensson,Journal of the American Chemical Society, 2003. 125(12): p. 3503-3508.
34. Deeth, R.J., A. Smith, and J.M. Brown,Journal of the American Chemical Society, 2004. 126(22): p. 7144-7151.
35. Ludwig, M., et al.,Organometallics, 1999. 18(6): p. 970-975.
36. Nilsson, K. and A. Hallberg,Journal of Organic Chemistry, 1992. 57(14): p.
4015-4017.
37. Ripa, L. and A. Hallberg,Journal of Organic Chemistry, 1997. 62(3): p. 595-602.
38. Jeffery, T.,Tetrahedron Letters, 1991. 32(19): p. 2121-2124.
39. Larock, R.C. and W.H. Gong,Journal of Organic Chemistry, 1990. 55(2): p. 407-408.
40. Nilsson, K. and A. Hallberg,Journal of Organic Chemistry, 1990. 55(8): p. 2464-2470.

